Breakout room 2: BLAST reprise, and RNA sequencing pipeline
Overview
Teaching: 0 min
Exercises: 30 minQuestions
Objectives
Re-run containerised BLAST with a custom database location
Execute a RNA sequencing pipeline in a container
Goal 1: BLAST reprise
In this first exercise, you’re going to repeat some of the steps of the previous BLAST exercise, with a key difference: this time, the reference database will not be located in the current working directory, but in a custom, distinct path somewhere else in the directory tree. You will then need to explicitly bind mount that path to be able to BLAST the sequence about it.
Start with cding into the exercise directory, containing the sequence to be BLASTED:
cd /data/bio-intro-containers/exercises/blast_2
ls *.fasta
P04156.fasta
Note that the reference database is in a distinct path, /data/bio-intro-containers/exercises/database_blast:
ls ../database_blast
zebrafish.1.protein.faa.gz
Guided preparation
You can re-use the same BLAST image you already downloaded. To save time in typing (and reduce typos!), you can store the full path in a shell variable for later use:
export blast_image="/data/bio-intro-containers/exercises/blast_1/blast_2.9.0--pl526h3066fca_4.sif"
Check this all works:
ls $blast_image
blast_2.9.0--pl526h3066fca_4.sif
Now cd into the database directory and prepare the database once again:
cd ../database_blast
gunzip zebrafish.1.protein.faa.gz
singularity exec $blast_image makeblastdb -in zebrafish.1.protein.faa -dbtype prot
Building a new DB, current time: 11/16/2019 19:14:43
New DB name: /data/bio-intro-containers/exercises/database_blast/zebrafish.1.protein.faa
New DB title: zebrafish.1.protein.faa
Sequence type: Protein
Keep Linkouts: T
Keep MBits: T
Maximum file size: 1000000000B
Adding sequences from FASTA; added 52951 sequences in 1.34541 seconds.
And finally cd back to the directory with the sequence file:
cd ../blast_2
Now run the alignment
This is the alignment command:
blastp -query P04156.fasta -db ../database_blast/zebrafish.1.protein.faa -out results.txtIf you run it as you did in the previously exercise, you will get a file not found error:
Error
singularity exec $blast_image blastp -query P04156.fasta -db ../database_blast/zebrafish.1.protein.faa -out results.txtBLAST Database error: No alias or index file found for protein database [../database_blast/zebrafish.1.protein.faa] in search path [/data/work/gitrepos/Trainings/bio-intro-containers/exercises/blast_2::]Then, try and run the alignment command from the container, while also bind mounting the directory containing the database using appropriate Singularity syntax.
Solution 1
singularity exec -B ../database_blast $blast_image blastp -query P04156.fasta -db ../database_blast/zebrafish.1.protein.faa -out results.txtSolution 2
export SINGULARITY_BINDPATH="../database_blast" singularity exec $blast_image blastp -query P04156.fasta -db ../database_blast/zebrafish.1.protein.faa -out results.txtMore possible solutions
Note that the full path of the database directory is
/data/bio-intro-containers/exercises/database_blast.
Therefore, mounting any of these would also work:/data,/data/bio-intro-containers,/data/bio-intro-containers/exercises.
The final results are stored in results.txt:
Goal 2: RNA sequencing pipeline
In this exercise, you’re going to port a small RNA sequencing pipeline to containers.
This is based on the RNAseq-NF repository by the Nextflow developers. Here is a graphical representation:
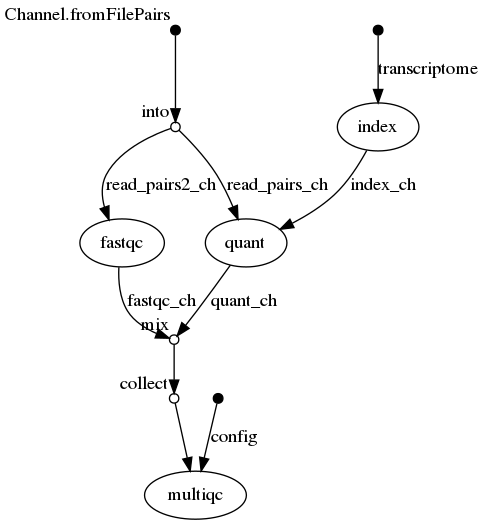
The pipeline uses three tools, salmon, fastqc and multiqc.
By containerising it, you’re going to further apply some skills you have just learnt about:
- download images;
- execute commands in containers through;
- bind mount additional host directories using one of:
- execution flag
-B; - environment variable
SINGULARITY_BINDPATH.
- execution flag
To save you some potentially long download times, we have pre-cached for you the required container images required by this workshop, so that pull commands will only take a few seconds. To take advantage of this, it’s important that you use the suggested images specifications.
Now, start with changing directory to the one for this exercise:
cd /data/bio-intro-containers/exercises/pipeline/data
It contains input files and useful scripts:
ls
clean_outputs.sh ggal_gut_1.fq ggal_gut_2.fq images.sh original_pipe.sh solutions
Use two terminal windows if you can
If you can, open at least two terminal windows, and connect to the VM from both of them. In this way, you can use one to edit files, and one to execute commands, thus making your workflow more efficient.
Preparation: have a look at the pipeline
Let’s have a look at the pipeline with cat:
cat original_pipe.sh
So here is the contents:
#!/bin/bash
echo "Pipeline started..."
# step 1
cd ../reference
# ONLY CHANGE THE NEXT LINE - EXECUTION LINE
salmon index -t ggal_1_48850000_49020000.Ggal71.500bpflank.fa -i out_index &>log_index
cd ../data
echo " indexing over"
# step 2
# ONLY CHANGE THE NEXT LINE - EXECUTION LINE
salmon quant --libType=U -i ../reference/out_index -1 ggal_gut_1.fq -2 ggal_gut_2.fq -o ggal_gut &>log_quant
echo " quantification over"
# step 3
mkdir -p out_fastqc
# ONLY CHANGE THE NEXT LINE - EXECUTION LINE
fastqc -o out_fastqc -f fastq -q ggal_gut_1.fq ggal_gut_2.fq &>log_fq
echo " quality control over"
# step 4
mkdir -p out_multiqc
cd out_multiqc
ln -s ../ggal_gut .
ln -s ../out_fastqc .
# ONLY CHANGE THE NEXT LINE - EXECUTION LINE
multiqc -v . &>../log_mq
cd ..
echo " multiple quality control over"
echo "Pipeline finished. Outcomes not checked, up to you."
You can see there’s some printing here are there using echo, just to monitor the progress at runtime.
Some housekeeping is also being done, such as changing directories and creating output directories.
The key execution lines are just four though, and make use of three packages (let’s stick to these versions, they have been tested for this session, and proven to work):
- salmon 1.2.1
- fastqc 0.11.9
- multiqc 1.9
Pull the container images
To save time, you’re not going to search for the images on the online registry.
Instead, your first task is to download the following container images.IMPORTANT: use only these images, as they have been pre-cached in the virtual machine to speed up the pull processes.
quay.io/biocontainers/salmon:1.2.1--hf69c8f4_0 quay.io/biocontainers/fastqc:0.11.9--0 quay.io/biocontainers/multiqc:1.9--pyh9f0ad1d_0Solution
singularity pull docker://quay.io/biocontainers/salmon:1.2.1--hf69c8f4_0 singularity pull docker://quay.io/biocontainers/fastqc:0.11.9--0 singularity pull docker://quay.io/biocontainers/multiqc:1.9--pyh9f0ad1d_0
SIF images in your current directory
We’re in
/data/bio-intro-containers/exercises/pipeline/data.
Check the downloaded image files (.sifextension) are actually in the current directory, usingls.Solution
ls *.siffastqc_0.11.9--0.sif multiqc_1.9--pyh9f0ad1d_0.sif salmon_1.2.1--hf69c8f4_0.sif
Containerise the pipeline
Now it’s the time to edit the pipeline script and make it work with containers!
Make a copy of the original pipeline to work with:
cp original_pipe.sh pipe.1.sh
You will need to modify ONLY FOUR LINES of this file, namely the ones marked with the comment # ONLY CHANGE THE NEXT LINE - EXECUTION LINE. Do not change anything else, nor move files around in the directory structure.
Hints
- both
nanoandvitext editors are available, use whichever you prefer; - add
singularitysyntax to execute those four commands using the appropriate containers you downloaded; - the current directory is mounted by default in the container;
- if you need other directories, however… (to bind mount use the execution flag approach for now, not the environment variable);
- if you want to go in stages, you can temporarily add an
exitcommand in the script after the pipeline step you’re working on, to avoid errors at the upcoming steps (that are not yet containerised); - if you need to clean up the outputs in the work directories to perform further tests, you can use the provided script
clean_outputs.sh.
Step 1 - salmon index
Only the relevant edited line is reported.
Backslashes\and newlines are used for the sake of readability of the code.Hint: look in the script for the current directory for this step; you’ll need to use an appropriate path for the container image.
Solution
singularity exec \ ../data/salmon_1.2.1--hf69c8f4_0.sif \ salmon index -t ggal_1_48850000_49020000.Ggal71.500bpflank.fa -i out_index &>log_index
Step 2 - salmon quant
Hint: look at the script. Where are the reference data compared to working directory of this step?
Solution
singularity exec \ -B ../reference \ ./salmon_1.2.1--hf69c8f4_0.sif \ salmon quant --libType=U -i ../reference/out_index -1 ggal_gut_1.fq -2 ggal_gut_2.fq -o ggal_gut &>log_quant
Step 3 - fastqc
Solution
singularity exec \ ./fastqc_0.11.9--0.sif \ fastqc -o out_fastqc -f fastq -q ggal_gut_1.fq ggal_gut_2.fq &>log_fq
Step 4 - multiqc
Hint: look at the script. Where are the relevant input files/data compared to the current directory?
Solution
singularity exec \ -B .. \ ../multiqc_1.9--pyh9f0ad1d_0.sif \ multiqc -v . &>../log_mq
Whenever you’re happy with your updated pipeline script, run it:
./pipe.1.sh
It should take under a minute. If it runs to completion, among others you should have the output file out_multiqc/multiqc_report.html , about 1.2 MB in size:
ls -lh out_multiqc/multiqc_report.html
-rw-r--r-- 1 ubuntu ubuntu 1.2M Jun 8 13:01 out_multiqc/multiqc_report.html
You’ve just reflected on how to containerise an entire pipeline, well done!
Entire script solution
Here is the entire solution (
solutions/pipe.1.sh):Solution
#!/bin/bash echo "Pipeline started..." # step 1 cd ../reference # ONLY CHANGE THE NEXT LINE - EXECUTION LINE singularity exec \ ../data/salmon_1.2.1--hf69c8f4_0.sif \ salmon index -t ggal_1_48850000_49020000.Ggal71.500bpflank.fa -i out_index &>log_index cd ../data echo " indexing over" # step 2 # ONLY CHANGE THE NEXT LINE - EXECUTION LINE singularity exec \ -B ../reference \ ./salmon_1.2.1--hf69c8f4_0.sif \ salmon quant --libType=U -i ../reference/out_index -1 ggal_gut_1.fq -2 ggal_gut_2.fq -o ggal_gut &>log_quant echo " quantification over" # step 3 mkdir -p out_fastqc # ONLY CHANGE THE NEXT LINE - EXECUTION LINE singularity exec \ ./fastqc_0.11.9--0.sif \ fastqc -o out_fastqc -f fastq -q ggal_gut_1.fq ggal_gut_2.fq &>log_fq echo " quality control over" # step 4 mkdir -p out_multiqc cd out_multiqc ln -s ../ggal_gut . ln -s ../out_fastqc . # ONLY CHANGE THE NEXT LINE - EXECUTION LINE singularity exec \ -B .. \ ../multiqc_1.9--pyh9f0ad1d_0.sif \ multiqc -v . &>../log_mq cd .. echo " multiple quality control over" echo "Pipeline finished. Outcomes not checked, up to you."
Bonus: alternate script with containers
If you want, have a look at this alternate solution (
solutions/pipe.2.sh).It solves the same requests than above, with 2 differences that improve the clarity and readability of the script:
- image paths are stored in variables at the beginning;
- bind mounts are done at the beginning using the variable
SINGULARITY_BINDPATH.Solution
#!/bin/bash salmon_image="/data/bio-intro-containers/exercises/pipeline/data/salmon_1.2.1--hf69c8f4_0.sif" fastqc_image="/data/bio-intro-containers/exercises/pipeline/data/fastqc_0.11.9--0.sif" multiqc_image="/data/bio-intro-containers/exercises/pipeline/data/multiqc_1.9--pyh9f0ad1d_0.sif" export SINGULARITY_BINDPATH="/data" echo "Pipeline started..." # step 1 cd ../reference # ONLY CHANGE THE NEXT LINE - EXECUTION LINE singularity exec \ $salmon_image \ salmon index -t ggal_1_48850000_49020000.Ggal71.500bpflank.fa -i out_index &>log_index cd ../data echo " indexing over" # step 2 # ONLY CHANGE THE NEXT LINE - EXECUTION LINE singularity exec \ $salmon_image \ salmon quant --libType=U -i ../reference/out_index -1 ggal_gut_1.fq -2 ggal_gut_2.fq -o ggal_gut &>log_quant echo " quantification over" # step 3 mkdir -p out_fastqc # ONLY CHANGE THE NEXT LINE - EXECUTION LINE singularity exec \ $fastqc_image \ fastqc -o out_fastqc -f fastq -q ggal_gut_1.fq ggal_gut_2.fq &>log_fq echo " quality control over" # step 4 mkdir -p out_multiqc cd out_multiqc ln -s ../ggal_gut . ln -s ../out_fastqc . # ONLY CHANGE THE NEXT LINE - EXECUTION LINE singularity exec \ $multiqc_image \ multiqc -v . &>../log_mq cd .. echo " multiple quality control over" echo "Pipeline finished. Outcomes not checked, up to you."
Key Points
Bind mount additional directories in the container with the flag
-B, or the variableSINGULARITY_BINDPATHContainerise an entire pipeline by making use of
singularity exec <image>and few other markups